Separase as a trigger of intrinsic apoptosis
Introduction
The intrinsic pathway of apoptosis is regulated by a balance between pro- and anti-apoptotic Bcl2 family proteins (e.g. Bak, Bax, Bcl2 itself, Bcl-xL, Mcl1, Bim and Bad) that are hallmarked by presence of one to four Bcl2 homology (BH) domains. Pore formation by homo-oligomerization of Bak and Bax leads to mitochondrial outer membrane permeabilization (MOMP) and release of cytochrome c and other apoptogenic factors from the intermembrane space. MOMP is counteracted by family members like Bcl2 itself, Bcl-xL and Mcl1. They employ a hydrophobic groove formed by their BH1-3 domains to sequester the BH3 domain of Bak/Bax and inhibit their self-interaction. BH3-only proteins like Bim or Bad activate Bak/Bax either directly by transient interaction or indirectly by competing Bak/Bax off anti-apoptotic Bcl2 members. Excessive cellular stress, like overwhelming DNA damage, triggers the intrinsic pathway of apoptosis. Most studies on this topic find that a hyperactive DNA damage response results in phosphorylation-dependent stabilization of p53 and its activation as a transcription factor. Consequent expression of pro-apoptotic target genes then results in a surplus of BH3-only proteins, which tips the balance towards MOMP and consequent cell death.
Whether intrinsic apoptosis can also be triggered in a transcription-independent manner is much less studied and incompletely understood. Likewise, it remains unclear whether proteins other than BAK/BAX might also be able to form pores and contribute to MOMP.
Separase enforces minimal length of mitosis
Securin and Sgo2-Mad2 are the major inhibitors of human separase. Co-depletion of securin and Sgo2 suffered not only from premature loss of cohesion but, unexpectedly, also from death in mitosis (DiM). Further investigations revealed that cells, which enter mitosis with already active separase, suffer from separase-dependent cleavage of the survival factors Mcl1 and Bcl-xL. This not only releases from their grip pro-apoptotic Bak and Bax but at the same time also transforms Mcl1 and Bcl-xL themselves into apoptogenic fragments. Importantly, the deadliest cleavage fragment, the C-terminal half of MCL1, forms BAK/BAX-like pores in the mitochondrial outer membrane.
Why are Mcl1 and Bcl-xL not cleaved in an unperturbed cell, in which separase is activated on schedule, i.e. at the end of metaphase? As it turned out, both anti-apoptotic factors serve as separase substrates only when phosphorylated by Nek2a, and this kinase is degraded already during prophase. (In case of Bcl-xL, Cdk1/2-cyclin A2-dependent phosphorylation contributes to cleavability but cyclin A2 exhibits the same early mitotic degradation as Nek2a.) Thus, cells do not kill themselves at anaphase onset because, by then, Mcl1 and Bcl-xL are dephosphorylated and no longer recognized by separase as substrates. In search for a biological function of separase-triggered DiM, we discovered that the rapid traverse of mitosis by cells with a compromised SAC results in activation of separase before all Nek2a is gone. Indeed, this temporal overlap of the two enzymatic activities resulted in cleavage of Mcl1 and Bcl-xL followed by intrinsic apoptosis. Thus, Nek2a and separase together represent a reactive cellular clock which supervises the length of early mitosis. Consequently, cells, which rush through this crucial cell cycle phase and will therefore likely suffer from chromosome missegregation, are eliminated by programmed cell death. Given that chromosomal instability is a driver (and possible cause) of malignancies, this might protect the organism from cancer.
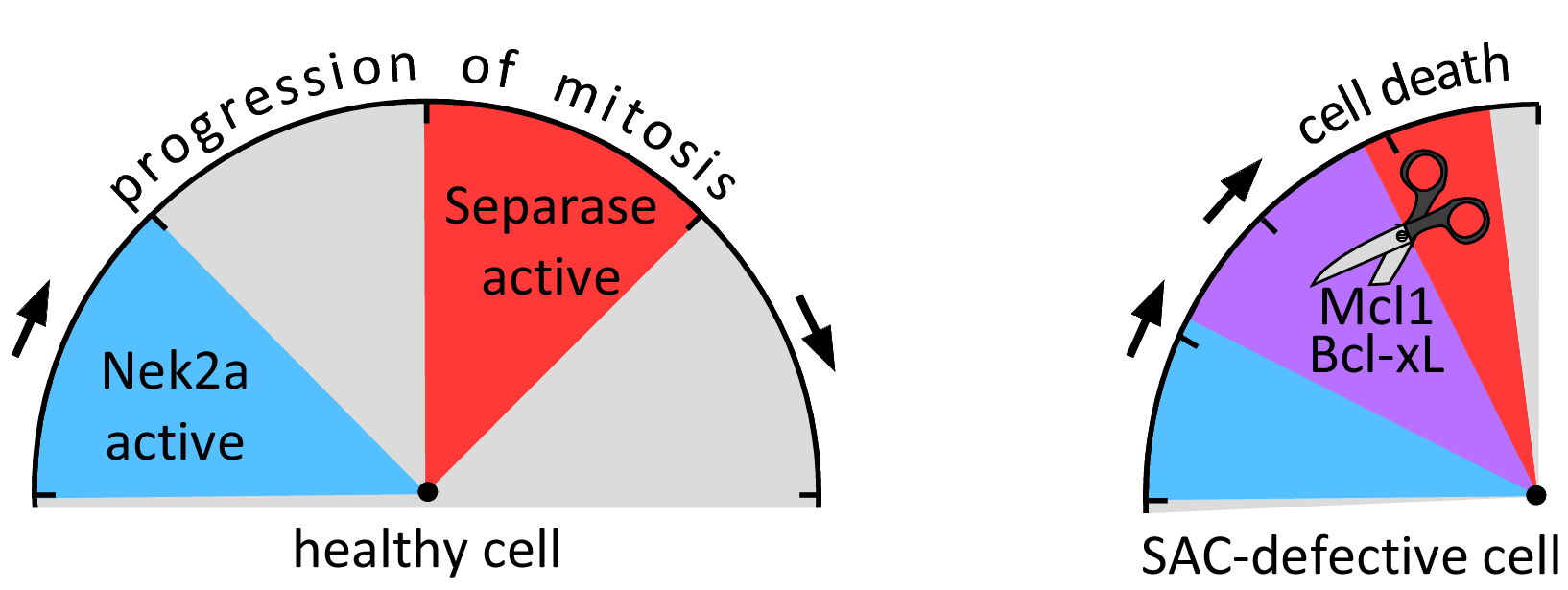
Fig. 4: Together with Nek2a, Separase constitutes a reactive cellular clock that induces intrinsic apoptosis if mitosis falls below a minimal duration.