Sister chromatid cohesion and separation
Introduction
After replication, each chromosome consists of two identical sister chromatids, which are topologically embraced by cohesin, a ring-shaped protein complex. Sister chromatid cohesion is essential for later, accurate halving of the genetic material. However, before chromosome segregation can take place, cohesion needs to be resolved. In metazoans, this occurs in two waves. The concerted actions of protein kinases and Wapl open cohesin rings in a non-proteolytic manner. While this so-called prophase pathway removes cohesin from chromosome arms, cohesin at centromeres is protected by Sgo1-mediated dephosphorylation. Activation of separase by ubiquitin-dependent degradation of its inhibitor securin results in cleavage of the Rad21 subunit of remaining cohesin. It is this proteolysis-dependent, second wave of ring opening, which universally triggers eukaryotic anaphase.
Although our grasp of chromosome cohesion and segregation has greatly advanced since the late 1990s, many questions remain to be answered. The prophase pathway has not been reconstituted to date, thus leaving unanswered whether all relevant kinases and their substrates have been identified? Unclear is also how Sgo1 is (transiently) targeted to centromeric cohesin, why it leaves again well before anaphase onset, and why this does not result in Wapl-dependent, premature loss of sister centromere cohesion? And while it is clear that securin is not essential, many issues of separase regulation in its absence remain to be unraveled.
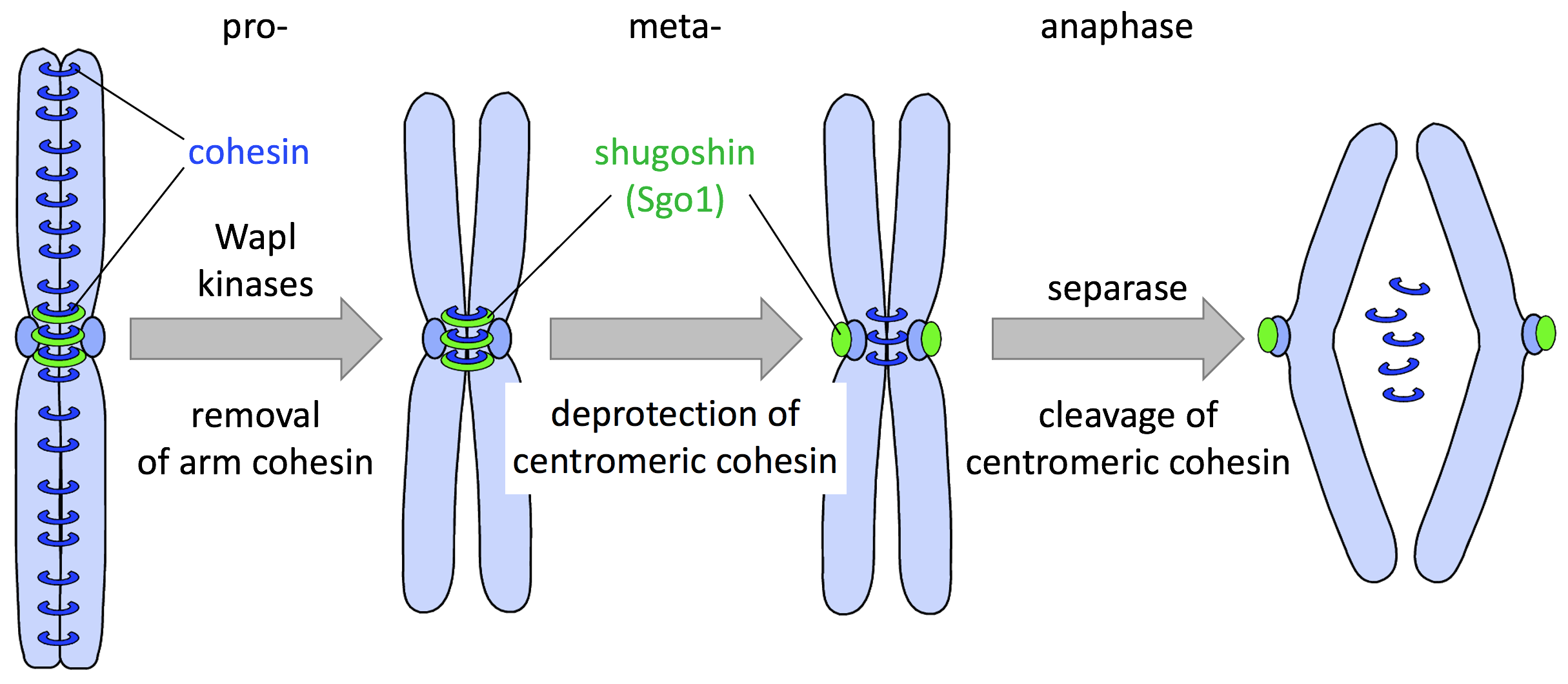
Fig. 1: The stepwise separation of sister chromatids in metazoan mitosis.
Mutual regulation of separase and securin
Prior to anaphase, separase is held inactive by association with securin, which blocks separase's active site with a non-cleavable pseudo-substrate sequence. While it was clear that both proteins are co-regulated and positively influence each other, the underlying mechanisms remained unknown. We could show that securin has no impact on the level of separase mRNA but associates co-translationally with separase. Thereby, it likely assists separase to adopt a proper native fold, from which it can later be activated by destruction of securin. Vice versa, securin's half-life is increased by binding to separase. This is because phosphorylation of free human securin speeds up its ubiquitin-dependent degradation, while separase, by recruiting protein phosphatase 2A (PP2A), keeps associated securin in a de-phosphorylated state. This preferred destruction of free over separase-bound securin may sharpen the metaphase-to-anaphase transition.
Regulation of separase by Cdk1-cyclin B1
Surprisingly, the loss of securin leaves mice and cultured human cells largely unaffected and raised the question of how separase is regulated in the absence of this anaphase inhibitor? Addressing this issue, we previously identified the master regulatory kinase of mitosis, Cdk1-cyclin B1, as a securin-independent inhibitor of separase. Formation of the separase-Cdk1-cyclin B1 complex requires prior Cdk1-dependent phosphorylation of separase followed by Pin1-catalyzed peptidyl-prolyl cis/trans isomerization. Andreas Boland and coworkers recently determined the structure of the human separase-Cdk1-cyclin B1 complex by cryo-EM. Interestingly, it shows that, in contrast to securin, which bears a pseudo-substrate sequence on its own, the kinase orients an otherwise disordered loop of separase to now act as an auto-inhibitory pseudo-substrate. Curiously, the same loop also blocks the active site of the kinase, which is in line with our previous finding that separase is not only a protease but also a Cdk1 inhibitor.
In a wild type metaphase cell most separase is in complex with securin and only a small fraction with Cdk1-cyclin B1. (Both inhibitors bind separase in a mutually exclusive manner.) Yet, securin-associated separase is still phosphorylated by Cdk1 and, upon degradation of securin (and cyclin B1), undergoes Pin1-dependent isomerization. We found that this has three functional consequences: It renders separase resistant to re-inhibition by any residual securin that escaped destruction. At the same time, it sensitises separase to (irreversible) inactivation by aggregation/misfolding and to (reversible) association with Cdk1-cyclinB1, which is additionally promoted by dephosphorylation of residual cyclin B1 that has not yet been destroyed. As a result, a short burst of separase activity is followed by a second peak of separase-Cdk1-cyclin B1 formation in late M-phase. This ensures that cohesin can be re-loaded onto chromatin in telophase without being cleaved. In addition, separase-dependent inhibition of the last Cdk1 might facilitate cytokinesis. Furthermore, the final degradation of the very last cyclin B1 releases a small pool of active separase in early G1 phase, which then triggers centriole disengagement, thereby licensing later centrosome duplication.
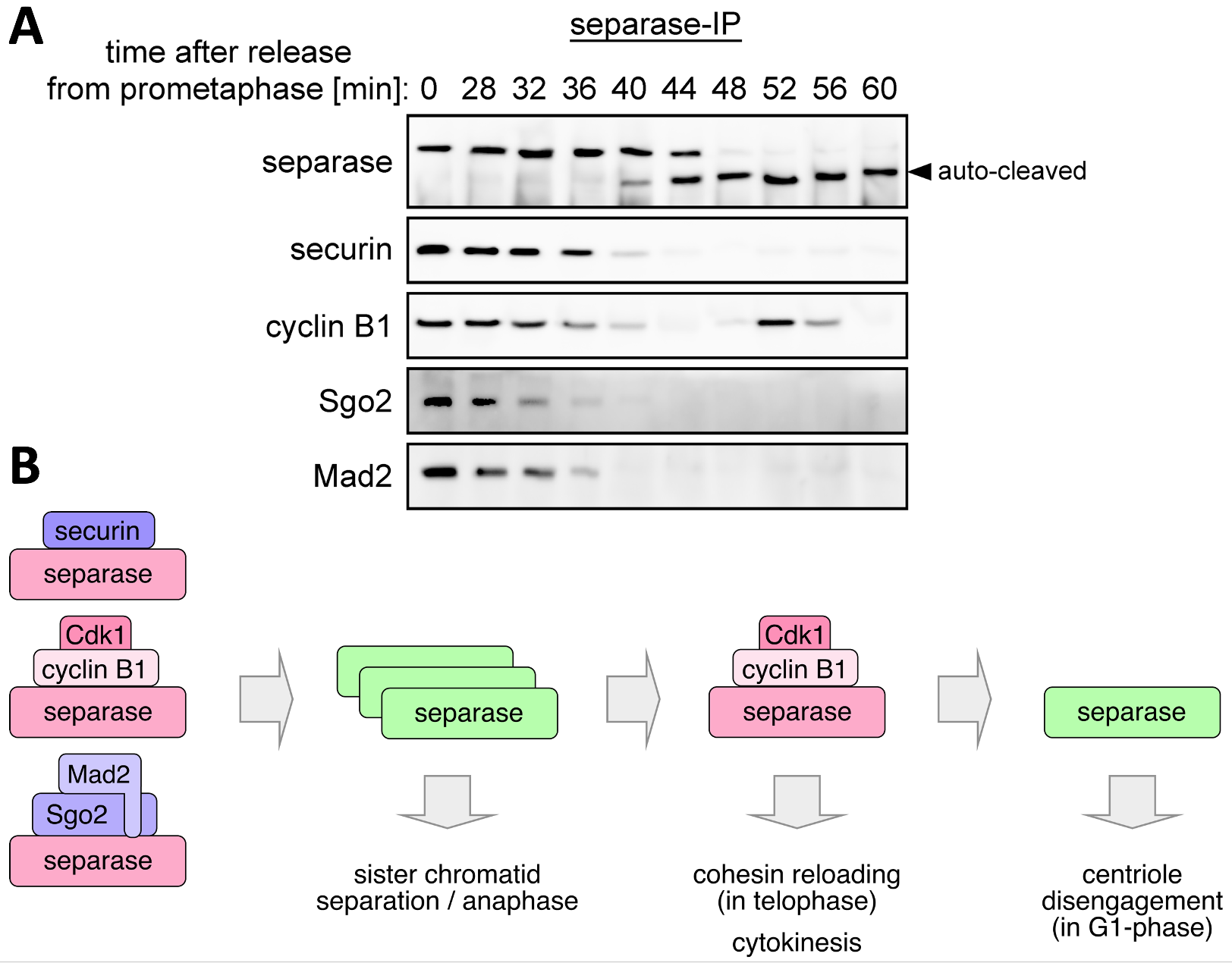
Fig. 2: Complexes and activities of separase from prometa- until early G1-phase. A) Time-resolved immunoprecipitation (IP) of separase from cells going synchronously through M-phase and into G1. Associated proteins were analyzed by immunoblotting. B) Model of the data from A). Red = inhibited (separase or Cdk1); green = active (separase).
Inhibition of separase by checkpoint-induced Sgo2–MAD2
With Sgo2 we recently discovered a 3rd inhibitor of human separase, which operates independent of (and parallel to) securin and Cdk1-cyclinB1. Sgo2 inhibits separase in a securin-like, competitive manner, i.e. by using a pseudosubstrate motif to block the protease's active site. Curiously, inhibition of separase by Sgo2 requires active spindle assembly checkpoint (SAC) signalling. The SAC is a surveillance mechanism that creates a diffusible 'wait anaphase signal' as long as chromosomes do not yet properly interact with the mitotic spindle apparatus. More specifically, the SAC catalyzes at un- or misattached kinetochores the conformational activation of free Mad2 from an 'open' via a transitional state into a 'closed' conformation, which locks onto client proteins. Sequestration of Cdc20 by Mad2 blocks the ubiquitin-dependent degradation of securin and cyclin B1. In contrast, association of Mad2 with Sgo2 enables the latter to bind and inhibit separase. Once the SAC is silenced, the AAA-ATPase Trip13, in conjunction with its Mad2-specific adaptor p31comet, actively disassembles Mad2-containing complexes. This results in destruction of securin and cyclin B1 on the one hand and liberation of separase from the grip of Sgo2 on the other hand. These results not only identified an unexpected function of Sgo2 in mitotically dividing cells but unraveled also a mechanism of separase regulation that is independent of securin and Cdk1-cyclin B1 but still supervised by the SAC.