Hellmuth Group
Current Research Themes
Research Focus I: MDM2-dependent mitotic time-keeping and p53-mediated cell cycle arrest
My current research focuses on mechanisms that monitor mitotic progression and translate mitotic perturbations into defined cell cycle outcomes. Central to this work is the concept that cells actively monitor the duration and quality of mitosis and convert this information into a molecular signal that determines subsequent cell fate decisions. In particular, I investigate how MDM2-dependent regulatory processes function as a mitotic time-keeping mechanism that controls p53 activation and enforces a G1 arrest following aberrantly prolonged mitosis.
Using genetic, biochemical, and cell biological approaches, my research aims to define how mitotic timing information is generated, stabilized, and transmitted across cell cycle phases. A key goal is to understand how cells distinguish physiological mitosis from mitotic stress, how transient mitotic signals persist beyond mitotic exit, and how these signals interface with the p53 pathway to impose long-term cell cycle control.
By dissecting the molecular logic of MDM2-dependent mitotic surveillance, this project seeks to establish general principles by which proliferating cells integrate their mitotic history into genome stability maintenance and cancer-relevant cell fate decisions.
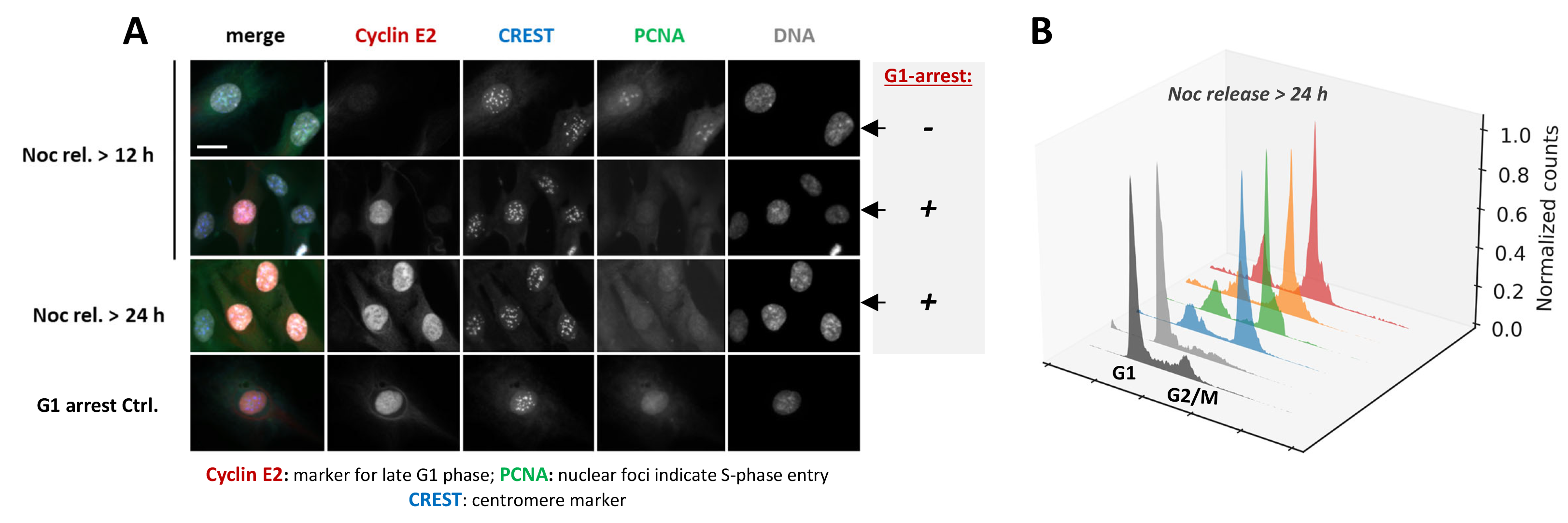
G1 arrest following mitotic delay. (A) Immunofluorescence analysis of human retinal pigment epithelial (RPE) cells 12 or 24 h after release from nocodazole-induced mitotic delay. Low-dose nocodazole treatment causes subtle spindle defects, thereby prolonging mitosis in a SAC-dependent manner and increasing the risk of chromosome segregation errors. Following drug removal, cells translate this mitotic delay into post-mitotic cell cycle control decisions. Cells that establish a stable p53-dependent G1 arrest and prevent replication of potentially damaged or missegregated chromosomes, are identified by Cyclin E2 expression (red). In contrast, cells that fail to maintain this arrest re-enter S phase, marked by PCNA foci formation (green). Representative images illustrate these distinct cell cycle outcomes after mitotic stress. (B) Flow cytometric analysis of propidium iodide–stained RPE cells 24 h after nocodazole washout. DNA content histograms illustrate cell cycle distributions of wild-type cells (black + grey plots), which predominantly accumulate in G1, in comparison to gene edited cell lines with impaired maintenance of the post-mitotic arrest that continue to cycle (blue, green, orange and red plots).
Research Focus II: Cell cycle–regulated DNA repair at centromeres and maintenance of centromeric chromatin
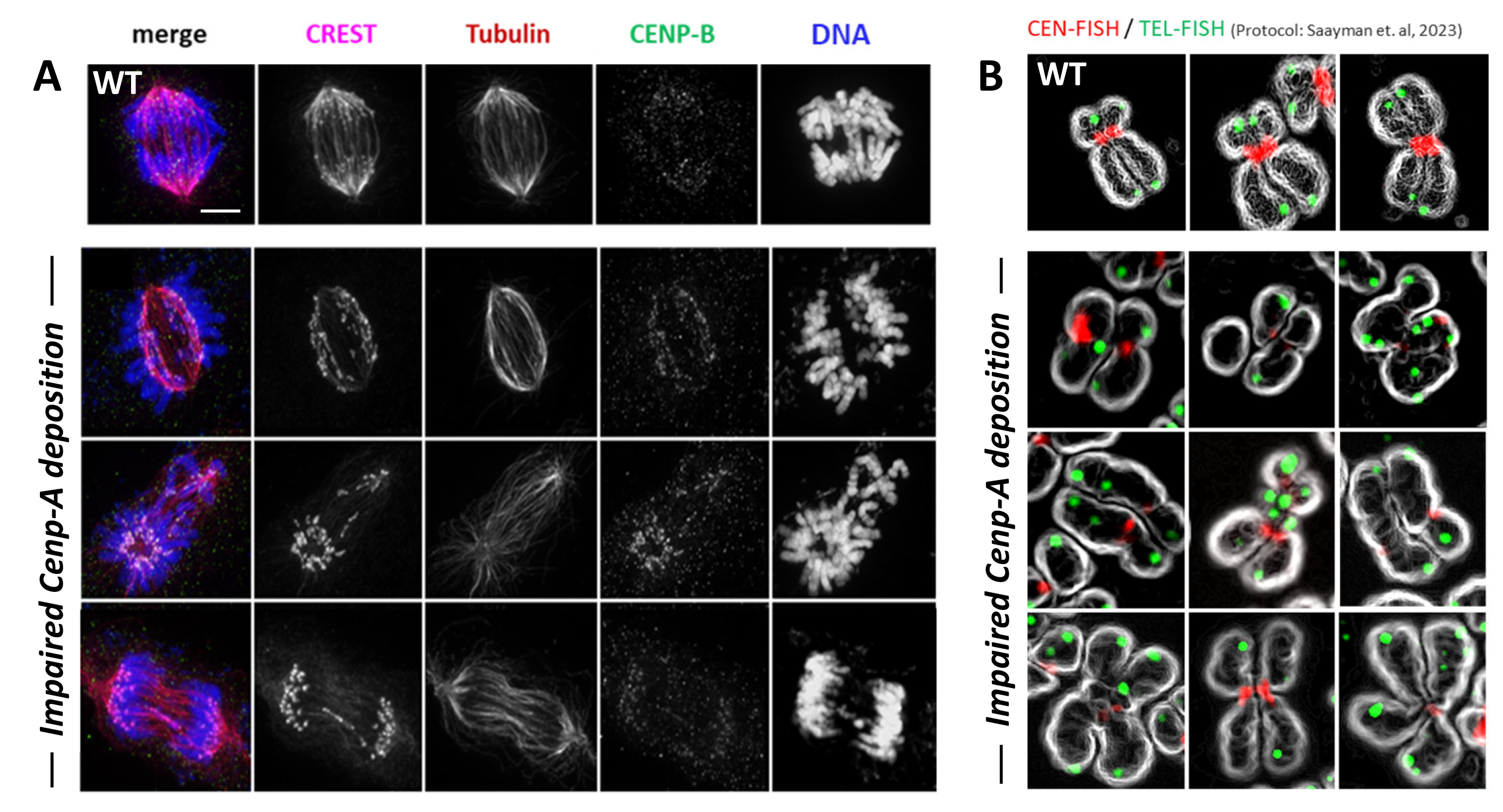
A second major research focus of my work addresses how DNA double-strand break (DSB) repair is regulated at centromeres and how repair pathway choice is adapted to the unique chromatin environment of these repetitive chromosomal regions. Centromeres are composed of alpha-satellite DNA assembled into CENP-A–containing nucleosomes and are inherently prone to replication stress and DNA damage, posing a particular challenge to genome integrity.
My research explores how cell cycle–dependent regulatory mechanisms locally modulate DNA repair factors at centromeres to permit homologous recombination-based repair outside its canonical temporal window. In this context, I am particularly interested in how regulatory attenuation of the DSB repair factor 53BP1 enables homologous recombination at centromeric satellite repeats, thereby supporting efficient repair and subsequent CENP-A loading.
This work highlights an unexpected functional coupling between mitotic regulatory pathways, DNA repair, and centromeric chromatin maintenance. Beyond repairing induced DNA damage, these mechanisms play a crucial role in preserving centromere identity and ensuring faithful chromosome segregation during normal cell cycle progression.
Together, this research aims to define how centromere-specific DNA repair pathways intersect with chromatin assembly processes to maintain genome stability under both physiological conditions and mitotic stress.
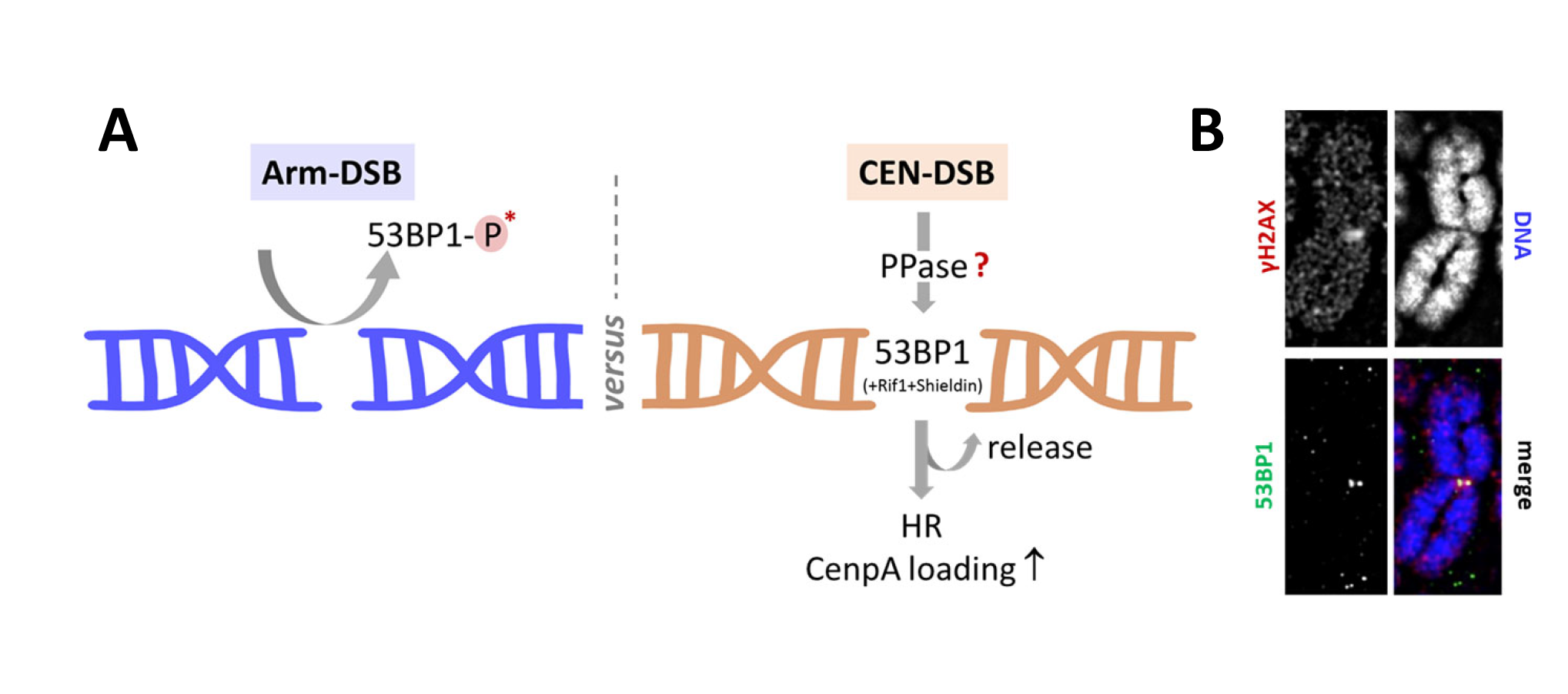
Schematic model for centromeric breakage and repair at the mitosis–G1 transition. (A) Proposed model illustrating cell cycle–dependent regulation of DNA double-strand break (DSB) repair at centromeres. During mitosis, DNA repair is globally suppressed, in part through inhibitory phosphorylation of DNA damage response factors, including 53BP1. At centromeres, however, local chromatin features and regulatory activities may permit site-specific modulation of 53BP1, allowing centromeric DSBs to be marked during late mitosis. I propose that local retention or reactivation of 53BP1 at centromeric breaks promotes recruitment of the anti–DNA end resection factor Shieldin, thereby biasing repair pathway choice towards NHEJ. Conversely, timely release of 53BP1 from centromeric damage sites may enable access of homologous recombination–associated factors such as BRCA1 and RAD51. This regulatory balance is proposed to influence repair outcome and to impact centromere maintenance, including CENP-A deposition at mitotic exit. (B) Metaphase chromosome spreads stained for the DNA damage marker γH2AX, illustrating centromeric damage signals and their spatial association with 53BP1